
FAQ
Here we collect frequent questions or issues which we try to answer or comment on. If your question is not here (yet) please don hesitate to contact us!
Samples will be put in queue when online sample submission has been performed and samples have been physically handed over to the facility. We hold on to the “first in, first sequenced” principle. Turn around times are:
- two weeks for RNA / DNA isolations
- three weeks for library preparation
- two weeks for sequencing run requests.
- two weeks for SNP fingerprinting
Be aware that turn around times depend on the number of requests, available reagents and personnel. Furthermore, we cannot control machine outage. In case of significant delays, we will contact you.
We are open for high priority requests. Note that we will discuss your request internally before granting.
DNA should be clean and isolated/purified with column base method. All concentrations MUST be measured with a proper fluorometric quantitation analysis (e.g. Qubit); Nanodrop is not reliable. If in doubt contact us. Samples that do not meet these criteria will cause severe delays in processing.
For ready-to-sequence samples:
- please deliver your (pooled) samples in an 1,5 ml tube with a concentration of 4 nM and a minimal volume of 40 ul.
For DNA library preparation:
- a minimum of 50 μl at a minimal concentration of 4 ng/μl.
- for (mammalian/plant) WGS: 500 ng/25 µl.
For RNA library preparation:
- RiboZero depletion: a minimum of 12 μl at a minimal concentration of 17 ng/μl.
- PolyA enrichment: a minimum of 50 μl at a minimal concentration of 4 ng/μl.
- RNA quality should be at least RIN 8 (RNA Integrity Number)
- We strongly advise to include DNase treatment during the RNA isolation process.
For Nanopore:
- DNA library prep: 1500 ng HMW in 48 µl MQ
- direct RNA: 500-1000 ng mRNA (!polyA) in 9 µl RNAsefree mq OR 75+ µg total RNA in 100 µl RNAseqfree mq
- CRISPR Cas9 Enrichment: 1-10 µg HMW DNA in 24 µl MQ
- please make sure DNA is free of proteins and/or small DNA fragments
High nucleotide diversity is when a library has roughly equal proportions of all 4 nucleotides in every cycle of the run. The diagram below illustrates the diversity and base-balance of well-balanced and unbalanced libraries, and how that can be reflected in the % base plot of Sequencing Analysis Viewer (SAV).
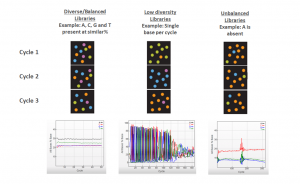
Why is nucleotide diversity important?
Nucleotide diversity is required for effective template generation on Illumina sequencing platforms and is important for the generation of high-quality data.
Diversity is especially important during the first 4–7 cycles of the first sequencing read for MiniSeq, MiSeq, NextSeq, and HiSeq 1000–2500 systems. The sequencing software uses images from these early cycles to identify the location of each cluster in a process called template generation.
Nucleotide diversity is also important for the first 25 cycles in the first sequencing read on all sequencing platforms because this is when phasing/pre-phasing, color matrix corrections, and the pass filter calculations occur. These corrections and calculations are used in base calling and quality score calculations for all cycles in a run for the clusters that pass filter.
The newest versions of Real-Time Analysis (RTA) software on the HiSeq 2500 and MiSeq platforms optimize the estimation of the color normalization matrix calculations and phasing/pre-phasing rates. These enhancements allow for low diversity and unbalanced libraries to be sequenced with a lower percentage of balanced library spike-in for color balance, resulting in higher quality sequencing data. When using HiSeq Control Software version 2.2.38 or higher, a minimum of 10% PhiX is required. When using MiSeq Control Software version 2.2 or higher, a minimum of 5% PhiX is required. If your sequencing platform is running a control software version lower than stated here, contact Illumina Technical Support for further guidance.
In systems running different versions of RTA, to achieve the same color balance, the input of anywhere between 10–50% PhiX may be required. For further guidance on the use of PhiX for color balance on other Illumina sequencing platforms, such as the MiniSeq and NextSeq, refer to the technical bulletins:
Best practices for low diversity sequencing on the NextSeq and MiniSeq systems
- Accurate and robust sequencing of low diversity libraries on the NextSeq and MiniSeq requires well-designed experiments and informatics pipelines. A common example of a low diversity library is an amplicon-based library preparation method, such as 16S metagenomics. These libraries tend to have DNA sequences that start at the same location – the probe binding site – and are mostly identical. A single represented locus causes a biased base composition that can change drastically from cycle to cycle.
The NextSeq and MiniSeq systems use 2-channel sequencing chemistry. Therefore, it is important to have all 4 DNA bases represented in every cycle, in order for the software to correctly identify DNA clusters and perform accurate base calling. To meet this requirement using a low diversity library, we recommend experimental designs that provide cycle-to-cycle diversity using the following methods: - Primer phasing
- Spike-in a whole-genome sample (eg, PhiX) to the run.
- A good starting point is to use 50% PhiX and then titrate the amount down, based on the quality of the primary and secondary analysis results. The presence of the spiked-in sample provides the necessary cycle-to-cycle base diversity.
With the exception of WGS, we do not support single sample submissions and we require samples sets to fill a a full flowcel or run. Why?
- avoid long waiting times. If we aim for a (most) cost-efficient single samples price, we have to collect a sufficient number of compatible samples to fill a run. In our experience, this is very unpredictable, leading to long waiting times.
- Very complex logistics and administration: if we would have sufficient samples collected for a full run, and during library prep samples drop out and need to be resubmitted, again there is a waiting time for the complete project.
- billing and accountancy issues: With a full run submission accountants can follow exactly what has been used for this project which is required to allow it on research grands.
- Prevention of cross influences of samples sets.